Documentation
Version 1.3 / 04.05.2018
Overview
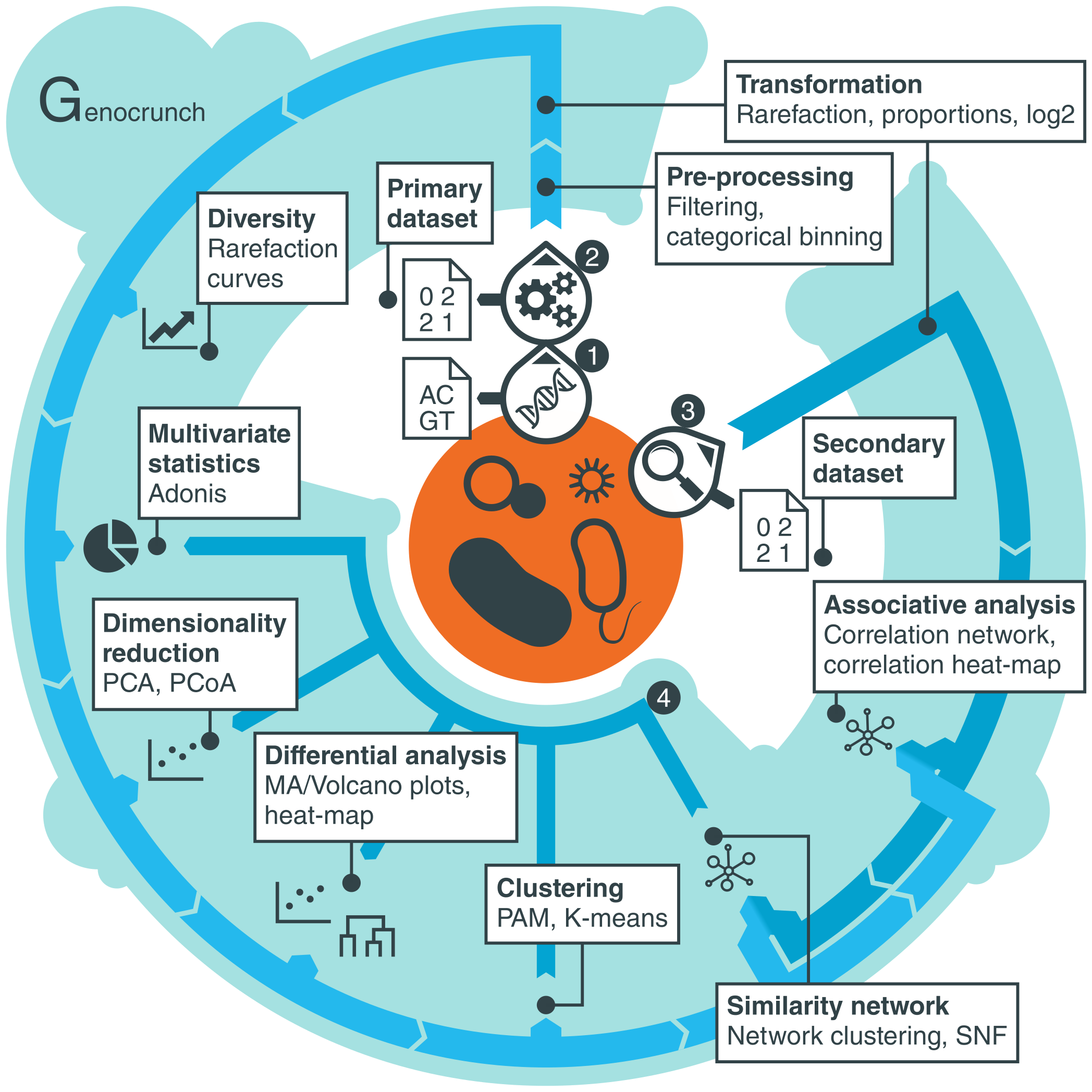
Genocrunch is a web-based data analysis platform dedicated to metagenomics and metataxonomics. It is tailored to process datasets derived from high-throughput nucleic acids sequencing of microbial communities  , such as gene counts or counts of operational taxonomic units (OTUs)
, such as gene counts or counts of operational taxonomic units (OTUs)  . In addition to such primary dataset, it also allows the integration of a secondary dataset (e.g. metabolites levels)
. In addition to such primary dataset, it also allows the integration of a secondary dataset (e.g. metabolites levels)  .
.
Genocrunch provides tools covering data pre-processing and transformation, diversity analysis, multivariate statistics, dimensionality reduction, differential analysis, clustering as well as similarity network analysis and associative analysis.
The results of clustering are automatically inferred as additional models into other analyses  .
.
Interactive visualization is offered for all figures.
New users and registration
Running an analysis session does not require any registration, however, managing multiple analysis (recovering/editing previous work) is done through a personnal index that requires users to sign in. Before signing in, new users need to register.
Registering
Registering is done via the Register button of the top-bar. The registration process requires new users to chose a password. The email address is only used to recover forgotten passwords.
Signing in
Signing in is done from the welcome page, which is accessible via the Sign In button of the top-bar. Before signing in, new users need to complete the registration process.
Trying Genocrunch
The best way to try Genocrunch is to load an example and edit it.
Examples
Examples of analyses are provided via the Examples link of the top-bar. Loading Example data into a new analysis session is possible via the Load button. This allows to visualize and edit Examples without restriction. For signed-in users, it will also copy the example into their personal analysis index.
Analyzing data
Running a new analysis
New analyses can be submitted via the New Analysis button of the topbar. If the user is signed out, the analysis session will be associated to the browser session and remain available as long as the browser is not closed. If the user had been signed-in, the analysis will be stored in his personal index. Analyses parameters are set by filling a single form comprising four parts for Inputs, Pre-processing, Transformation and Analysis. Analyses are then submitted via the Start Analysis button located at the bottom the form. See the Inputs and Tools section below for details.
Editing an analysis
Analyses can be edited via the associated Edit Analysis button present on the analysis page and via the edit icons () of the analysis index (for signed-in users only). Editing is performed by updating the analysis form and submiting the modifications via the Restart Analysis button located at the bottom of the form.
Cloning an analysis
Analyses can be copied via the copy icons () of the analysis index (for signed-in users only).
Deleting an analysis
Analyses can be deleted via the delete icons () of the analysis index (for signed-in users only). This process is irreversible.
Inputs and Tools
The analyses are set from a single form composed of four sections: Inputs, Pre-processing, Transformation, Analysis. These sections and their associated parameters are described below.
Inputs
The Inputs section of the analysis creation form allows to upload data files.
- General information
- Name (mandatory)
The name will appear in the analysis index. It should be informative enough to differentiate from other analyses.
- Description
The description can be used to provide additional information about the analysis.
- Name (mandatory)
- Data files
- Primary dataset (mandatory)
The primary dataset is the principal data file. It must contain a data table in the tab-delimited text format with columns representing samples and rows representing observations. The first row must contain the names of samples. The first column must contain the names of the observations. Both columns and rows names must be unique. A column containing a description of each observation, in the form of semi-column-delimited categories, can be added at the end. This format can be obtained from the BIOM format using the
biom_convertcommand.Format example:
#OTU ID Spl1 Spl2 Spl3 Spl4 taxonomy 1602 1 2 4 4 k__Bacteria; p__Actinobacteria; c__Actinobacteria; o__Actinomycetales; f__Propionibacteriaceae; g__Propionibacterium; s__acnes 1603 0 0 24 0 k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Clostridiaceae; g__Clostridium; s__difficile 1604 0 12 23 0 k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Enterococcaceae; g__Enterococcus; s__casseliflavus 1605 23 4 2 14 k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Veillonella; s__parvula 1606 45 10 42 12 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides; s__fragilis 1607 8 15 20 13 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae; g__Prevotella; s__copri
- Category column (mandatory)
To increase compatibility, the category column specifies which column in the pimary dataset contains a categorical description of the observations. This must be either the first or the last column.
- Map (mandatory)
The map contains information about the experimental design. It must contain a table in the tab-delimited text format with columns representing experimental variables and rows representing samples. The first column must contain the names of the samples as they appear in the primary dataset. The first row must contain the names of experimental variables. Both columns and rows names must be unique. This format is compatible with the mapping file used by the Qiime pipeline.
Format example:
ID Sex Treatment Spl1 M Treated Spl2 F Treated Spl3 M Control Spl4 F Control
- Secondary dataset
The secondary dataset is an optional data file containing additional observations. It must contain a data table in the tab-delimited text format with columns representing samples and rows representing observations. The first row must contain the names of samples as they appear in the primary dataset and the map. The first column must contain the names of the observations. Both columns and rows names must be unique.
Format example:
Metabolites Spl1 Spl2 Spl3 Spl4 metabolite1 0.24 0.41 1.02 0.92 metabolite2 0.98 0.82 1.12 0.99 metabolite3 0.05 0.11 0.03 0.02
- Primary dataset (mandatory)
Pre-processing
The Pre-processing section of the analysis creation form specifies modifications that will be applied to the primary dataset prior to analysis.
- Filtering
Data in the primary dataset can be filtered based on Relative or Absolute abundance and presence.
- Abundance threshold
Minimal abundance per sample to be retained.
- Presence thresholds
Minimal presence among samples to be retained.
Example: Filtering data with an absolute abundance threshold of 5 and and an absolute presence threshold of 2.
#Before filtering #OTU ID Spl1 Spl2 Spl3 Spl4 taxonomy 1602 1 2 4 4 k__Bacteria; p__Actinobacteria; c__Actinobacteria; o__Actinomycetales; f__Propionibacteriaceae; g__Propionibacterium; s__acnes 1603 0 0 24 0 k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Clostridiaceae; g__Clostridium; s__difficile 1604 0 12 23 0 k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Enterococcaceae; g__Enterococcus; s__casseliflavus 1605 23 4 2 14 k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Veillonella; s__parvula 1606 45 10 42 12 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides; s__fragilis 1607 8 15 20 13 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae; g__Prevotella; s__copri
#After filtering #OTU ID Spl1 Spl2 Spl3 Spl4 taxonomy 1604 0 12 23 0 k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Enterococcaceae; g__Enterococcus; s__casseliflavus 1605 23 4 2 14 k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Veillonella; s__parvula 1606 45 10 42 12 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides; s__fragilis 1607 8 15 20 13 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae; g__Prevotella; s__copri
- Abundance threshold
- Category binning
Data in primary dataset can be binned by category thanks to the category column.
- Binning levels
Category levels at which data should be binned.
- Category binning function
Chose whether binning should be performed by summing or averaging values.
Example: Binning data at level 2 with sum.
#Before binning #OTU ID Spl1 Spl2 Spl3 Spl4 taxonomy 1604 0 12 23 0 k__Bacteria; p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Enterococcaceae; g__Enterococcus; s__casseliflavus 1605 23 4 2 14 k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Veillonella; s__parvula 1606 45 10 42 12 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides; s__fragilis 1607 8 15 20 13 k__Bacteria; p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae; g__Prevotella; s__copri
#After binning ID Spl1 Spl2 Spl3 Spl4 taxonomy 1 23 16 25 14 k__Bacteria; p__Firmicutes 2 53 25 62 25 k__Bacteria; p__Bacteroidetes
- Binning levels
Transformation
The Transformation section of the analysis creation form propose additional modifications for both primary and secondary datasets.
- Rarefaction
Although controversial (McMurdie and Holmes, 2014), rarefaction is a popular method amongst microbial ecologists used to correct for variations in sequencing depth, inherent to high-throughput sequencing methods. It consists in random sampling (without replacement) of a fixed number of count from each sample to be compared. The same number of count being drawn out of each sample, this corrects for differences in sequencing depth while conserving the original count distribution among observations. Note that the analysis of diversity is particularly sensitive to variations in sequencing depth.
- Sampling depth
This specifies the number of count to be drawn from each sample.
Tip: The maximal sampling depth corresponds to the minimal sequencing depth. First check the sequencing depth of your data by suming the counts in each sample. Then chose a sampling depth accordingly.
- N samplings
This specifies how many times the random drawing should be repeated. If more than 1 random sampling is done, the result is the average of all random samplings.
- Sampling depth
- Transformation
Whether it is to correct for sequencing depth, to stabilize the variance or simply to improve the visualization of skewed data, a transformation step may be needed. Common methods include transforming counts into proportions (percent or counts per million) and applying a log. We are working to also propose more advanced transformations, including those developed for RNA sequencing (RNA-seq) as part of the DESeq2 (Love et al., 2014) and the Voom (Law et al., 2014) pipelines as well as methods to limit the effect of known experimental bias (or batch-effect), such as Combat.
- Transformation
Specifies which transformation method should be applied.
- Transformation
Analysis
The Analysis section of the analysis creation form sets which statistics to apply and which figures to generate.
- Experimental design
This sets the default statistical method to apply for the analysis. Two options are available: Basic and Advanced.
- Statistics (Advanced only)
Chose a statistical method to compare groups of samples. If Basic is chosen, this defaults to ANOVA.
- Model
Chose a model defining groups of samples. If Basic is chosen, a model can be picked among headers of the map. If Advanced is chosen, the model must be typed by the user in the form of an R formula. The formula must be compatible with the specified statistics. All terms of the formula must refer to column headers of the map.
- Simple design:
ID Subject Site Spl1 subject1 Hand Spl2 subject2 Hand Spl3 subject3 Hand Spl4 subject4 Foot Spl5 subject5 Foot Spl6 subject6 Foot
Statistics Model Comparing sites (parametric) T-test Site Comparing sites (non-parametric) Wilcoxon rank sum test Site - Simple paired design:
The order of paired samples in the map is important!
ID Subject Site Spl1 subject1 Hand Spl2 subject2 Hand Spl3 subject3 Hand Spl4 subject1 Foot Spl5 subject2 Foot Spl6 subject3 Foot
Statistics Model Comparing sites (parametric) Paired t-test Site Comparing sites (non-parametric) Wilcoxon signed rank test Site - Multiple comparisons:
ID Subject Site Spl1 subject1 Hand Spl2 subject2 Hand Spl3 subject3 Hand Spl4 subject4 Foot Spl5 subject5 Foot Spl6 subject6 Foot Spl7 subject7 Mouth Spl8 subject8 Mouth Spl9 subject9 Mouth
Statistics Model Comparing sites (parametric) ANOVA Site Comparing sites (non-parametric) Kruskal-Wallis rank sum test Site - Multiple comparisons with nesting:
ID Subject Site Spl1 subject1 Hand Spl2 subject2 Hand Spl3 subject3 Hand Spl4 subject1 Foot Spl5 subject2 Foot Spl6 subject3 Foot Spl7 subject1 Mouth Spl8 subject2 Mouth Spl9 subject3 Mouth
Statistics Model Comparing sites Friedman test Site | Subject - Additive model:
ID Treatment Gender Spl1 Treated M Spl2 Treated M Spl3 Treated M Spl4 Treated F Spl5 Treated F Spl6 Treated F Spl7 Control M Spl8 Control M Spl9 Control M Spl10 Control F Spl11 Control F Spl12 Control F
Statistics Model Assessing effects of treatment and gender ANOVA Treatment+Gender
- Statistics (Advanced only)
- Proportions
For each sample, display the proportions of each observation (as percent) in the form of a stacked barchart. This gives an overview of the primary dataset. This analysis is performed before applying any selected transformation.
- Diversity
Perform a diversity analysis of the primary dataset. This will display rarefaction curves for the selected diversity metric(s). Rarefaction curves are used to estimate the diversity in function of the sampling depth. A rarefaction curve showing an asymptotic behavior is considered as an indicator of sufficient sampling depth to observe the sample diversity. This analysis is performed before applying any selected transformation.
- metric
Chose a diversity metric. Choices include the richness as well as metrics included in the R vegan, ineq and fossil packages. The richness simply represents the number of different observations that are seen within a particular sample.
- Compare groups
If selected, diversity between groups will be assessed using statistics and model specified in the experimental design.
- metric
- perMANOVA
Perform a permutational multivariate analysis of the variance using the Adonis method from R package vegan. This method uses a distance matrix based on the primary dataset.
- Distance metric
Chose a distance metric. Choices include metrics available in the R vegan package as well as distances based on correlation coefficients.
- Model
For compatibility purpose, this model may differ from the model specified in the experimental design section. This model must be in the form of an R formula compatible with the Adonis function of the R vegan package. All terms of the formula must refer to column headers of the map.
- Strata
This is used to specify any nesting in the model. the strata must be compatible with the Adonis function of the R vegan package.
- Distance metric
- PCA
Perform a principal component analysis (PCA) on the primary dataset. PCA is a commonly used dimensionality reduction method that projects a set of observations onto a smaller set of components that capture most of the variance between samples.
- PCoA
Perform a principal coordinate analysis (PCoA) on the primary dataset. The PCoA is a form of dimensionality reduction based on a distance matrix.
- Distance metric
Chose a distance metric. Choices include metrics available in the R vegan package as well as distances based on correlation coefficients.
- Distance metric
- Heatmap
This displays the primary dataset on a heatmap of proportions. The columns (samples) and rows (observations) of the heatmap are re-ordered using a hierarchical clustering. Each observation will be individually compared between groups of samples based on the specified experimental design and associated p-values will be displayed on the side of the heatmap. Individual correlations between observations from the primary dataset and observations from the secondary dataset will also be displayed on the side of the heatmap when available. Samples will be color-coded according to the experimental design.
- Changes
This performs a differential analysis. Each observation will be individually compared between groups of samples based on the specified experimental design. Fold-change between groups as well as the associated p-values and mean abundance will be displayed on MA plots and volcano plots.
- Correlation network
This builds a network with nodes representing observations and edges representing the correlations. The network will be based on the primary dataset and will include the secondary dataset if available. Observations belonging to the primary and the secondary dataset will be represented using different node shapes. Nodes will be colored to include a number of additional information.
- Correlation method
Chose a correlation method. Choices include the Pearson correlation and the Spearman correlation.
- Correlation method
- Clustering
This applies a clustering algorithm to separate samples into categories. The categories will be added as a new column to the map. Categories will automatically be compared with ANOVA in relevant analysis.
- Algorithm
Chose a clustering algorithm. Choices include the k-means algorithm, the k-medoids algorithm (also known as the partitioning around medoids of pam algorithm) and versions of the k-medoids based on distance matrices.
- Algorithm
- Similarity network
This builds a network with nodes representing samples and edges representing a similarity score. If a secondary dataset is available, three networks will be build: one based on the primary dataset, one based on the secondary dataset and a third based on the fusion of these two networks. The similarity network fusion (SNF) is based on the R SNFtool package. A categorical clustering based on each network is applied.
- Metric for primary dataset
Chose the similarity metric to apply on the primary dataset.
- Metric for secondary dataset
Chose the similarity metric to apply on the secondary dataset.
- Clustering algorithm
Chose the clustering algorithm to assign samples to categories based on each similarity network.
- Metric for primary dataset
Versions
Genocrunch data analysis tools are based on R libraries. Used libraries are mentionned in the analysis report. The version of the libraries are available on the Versions page via the Infos menu of the topbar).
Analysis report
A detailed analysis report is generated for each analysis. It is available directly after creating/updating a new analysis via the My Analysis button of the topbar or via the corresponding icon () in the analysis index (for signed-in users only). The analysis report includes detailed descriptions of the tools and methods used, possible warning and error logs as well as intermediate files, final files and interactive figures as they become available. Information for the pre-processing and data transformation are available for the primary dataset and the optional secondary dataset in their respective sections. Figures and descriptions for each analysis are available in separate sections. If multiple binning levels were specified, figures for each levels can be accessed via the associated Level button.
Exporting figures and data
Figures and their corresponding legends and data can be exported via the associated Export menu.
- Standard export
Includes a vector image in the SVG format as well as a figure legend in HTML format. Data in a tab-delimited text format is also proposed for some figures.
- Advanced export
Includes raw data in JSON format. R-generated PDFs are also proposed for some figures.
Archive
Files containing initial, intermediate and final data as well as the analysis log and logs for standard output and standard error are automatically included into a .zip archive. This archive can be downloaded via the Download Archive button of the analysis report page or via the corresponding icon () in the analysis index (for signed-in users only). Although some figures may be automatically included in the archive in the form of a rough PDF file, the archive does generally not contain figure images. These should be exported separately via the Export menu.
Bugs report
During computation (while the analysis is running), the standard error (stderr) stream is redirected into a bugs report that can be downloaded using the Bugs Report button located at the bottom-right of the details page in the analysis report.
Usage limits
Time limits
Analyses are by default kept for 2 days folowing their last update. However, analyses owned by registered users are kept for 365 days folowing their last update. Old analyses are automatically deleted from the server after their reach these limits.
Storage limits
The storage quota is set to 0 Bytes per user.